 Intelligent Design
Intelligent Design
How the Eukaryotic Cell Cycle Is Controlled
Previously, I offered a short description of the eukaryotic cell cycle and its culmination with mitotic cell division. While the cell cycle is certainly highly complex, the processes that regulate its progression from one phase to the next are even more remarkable. For a simplified animation, see the video above. For general readers seeking a basic familiarity with the cell cycle, which always comes in handy, I recommend reviewing my previous article for context before continuing here.
The Role of Cyclin-Dependent Kinases in Cell Cycle Progression
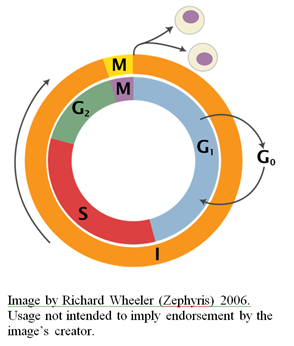
Progression of the eukaryotic cell cycle is driven by proteins called cyclin-dependent kinases (hereafter, Cdks). Cdks exert their effects by phosphorylating specific serine and/or threonine residues on their protein substrates that perform the various cell cycle events. Phosphorylation of protein substrates by Cdks can result in conformational changes that alter their catalytic activity and their interaction with other proteins. In humans, 292 CDK targets are known, and in the frog genus Xenopus, 77 targets have been identified (Errico et al., 2010). Many targets have also been identified in yeast (Enserink and Kolodner, 2010). These include proteins involved in DNA replication (Fisher, 2011; Mailand and Diffley, 2005; Frouin et al., 2002), chromosome condensation and mitosis (Bazile et al., 2010), and other processes that are a central part of cell cycle progression (Enserink and Kolodner, 2010).
Cdks are activated by the binding of regulatory proteins called cyclins to a conserved motif in the Cdk called PSTAIRE, to form cyclin-Cdk complexes.
The region of the cyclin protein that binds to the Cdk is about 100 amino acids in length and comprised of five alpha helices, referred to as the cyclin box (Noble et al., 1997; Lees and Harlow, 1993). Upon binding of the Cdk to its corresponding cyclin molecule, the Cdk undergoes a conformational change that renders the active site more accessible to its substrate.
Cdks can also promote the expression of the next cyclin, thereby facilitating the activation of the cyclin-Cdk complex of the next phase of the cell cycle. Cyclins are typically designated by the cell cycle phase that they control: Types of cyclins include G1 cyclins (Lew et al., 1991), G1/S cyclins (Moore et al., 1996), S cyclins (Dahmann et al., 1995) and M cyclins (Couchi et al., 1995). Cyclins are also grouped into different classes, per the table below.
| Cyclin-Cdk | Vertebrate Cyclins | Yeast Cyclins |
|---|---|---|
| G1 cyclin-Cdk | Cyclin D | Cln3 |
| G1/S cyclin-Cdk | Cyclin E | Cln1, 2 |
| S cyclin-Cdk | Cyclin A | Clb5, 6 |
| M cyclin-Cdk | Cyclin B | Clb1, 2, 3, 4 |
Different Cdks are activated at different cell stages by the specific cyclin molecules that are present at each respective stage. Cyclin levels rise at the beginning of a particular phase of the cell cycle, and subsequently undergo proteolytic degradation — leading to their abrupt decline — at the end of their specific phase (Glotzer et al., 1991). Indeed, the cyclical character of their accumulation and decline within the cell is what lends them their name. Take a look at the figure below (from here), representing the concentrations of different cyclin molecules throughout the cell cycle:
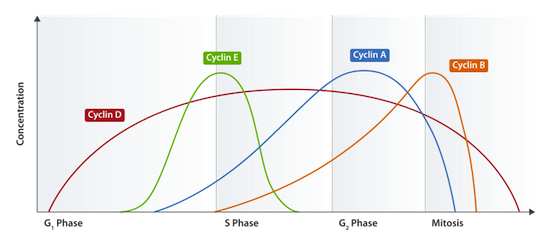
Cyclin-Cdks are also involved in regulating many of the cellular processes that are essential during the cell cycle, including disassembly of the nuclear envelope during mitosis (Guttinger et al., 2009). Phosphorylation by cyclin-Cdk furthermore controls changes in cell shape that occur in budding yeast during the cell cycle (Howell and Lew, 2012). Vacuole movement and reorganization of the Golgi apparatus during cell division are also controlled by cyclin-Cdk phosphorylation (Peng and Weisman, 2008; Millarte and Farhan, 2012).
{kind=link}
How Cyclin-Cdks Drive the Cell Cycle Forward
The network of interactions that push forward the cell cycle is extremely complex, involving many different proteins. To better understand the regulatory mechanism that drives cell cycle progression, consider by way of example the transition from S phase to M phase in budding yeast (which is unusual since there is no G2 phase in budding yeast, meaning that M phase immediately follows S phase). This transition requires the activity of M cyclin-Cdk. M cyclin, however, undergoes ubiquitin-mediated proteolysis prior to M phase, and so the S cyclin-Cdk (which drives S phase) is required to inhibit the proteolysis of M cyclin (Schwab et al., 1997). M cyclin thus accumulates and activates its corresponding Cdk (actually, in yeast, there only is one Cdk), thereby driving the initiation of M phase. The M cyclin-Cdk also acts to inhibit the transcription factors necessary for expression of the S phase cyclins, thereby putting a halt to the previous cell cycle phase.
Many of the target substrates of Cdks are transcription factors: For example, in mammals, transcription factor E2F is inhibited, prior to the passing through the G1 restriction point, by the binding of one of the Rb family of proteins (Calzone et al., 2008; Harbour and Dean, 2000). This means that it is not able to activate transcription. Rb dissociates from E2F when it is phosphorylated by a G1 cyclin-Cdk complex formed from the association between cyclin D and either Cdk4 or Cdk6. E2F is thus free to initiate the transcription of genes needed for beginning the cell cycle. Mutations in Rb can result in cancer development resulting from uncontrolled cell division (Nevins, 2000). In fact, Rb is itself named after a form of cancer, retinoblastoma, that results from mutations in that gene.
How Cyclin-Dependent Kinases Are Regulated by Phosphorylation
In addition to the regulation of Cdks by cyclin binding (which actually only causes partial activation), Cdks are also regulated by phosphorylation by carefully controlled enzymes called Cdk-activating kinases (hereafter, CAKs). Phosphate groups are also removed by phosphatase enzymes. The Cdk enzyme is activated by the phosphorylation of a specific threonine residue next to the active site.
In the case of M cyclin-Cdk, specific phosphate groups also need to be removed from the Cdk in order for full activation. There are two target sites of inhibitory phosphorylation of Cdks that are found in the ATP-binding site of the enzyme: One is at a specific tyrosine residue, and the other is at an adjacent threonine residue (Thr14 and Tyr15 in humans). Phosphorylation of these residues interferes with ATP orientation within the binding pocket, meaning that the M cyclin-Cdk is unable to phosphorylate its substrates.
The enzyme that phosphorylates these residues is a kinase called Wee1, which is active during G1, S, and G2 phases (Kellogg, 2003; Haese et al., 1995; Nurse, 1980). These phosphate groups are removed by a phosphatase enzyme called Cdc25, which becomes active in the G2 phase. In a remarkable feedback loop, both of these enzymes are themselves regulated by the M cyclin-Cdk complex, which is also their own substrate (Perry and Kornbluth, 2007). Upon M-phase cyclin Cdk phosphorylation, Cdc25 is activated and Wee1 is inhibited. The positive-feedback loop facilitates an abrupt transition from the G2 phase into mitosis due to the sharp increase in Cdk activity. In the event of DNA damage, Cdc25 is able to be inactivated by phosphorylation as a consequence of a pathway known as the DNA damage checkpoint pathway. When this happens, the Thr14/Tyr15 inhibitory phosphorylations are unable to be removed, and the cell cycle does not progress. I will discuss cell cycle check-point pathways in a future article.
Regulation by Cyclin Kinase Inhibitors
Activity of Cdks can also be inhibited by the binding of small proteins called cyclin kinase inhibitors (hereafter, CKIs), of which there are two classes. The CKIs of one class exert their effects by binding to a large domain on the Cdk, in addition to the hydrophobic patch of the cyclin, in G1-S or S phase cyclin-Cdk complexes. In doing so, the structure of the Cdk is distorted, and ATP binding is inhibited. CKIs of the other class bind instead to G1 Cdk monomers, causing a conformational change that alters the active site and reduces cyclin binding.
The budding yeast protein Sic1 is a CKI which exerts its inhibitory effects on the major S cyclin-Cdk1 and M cyclin-Cdk1 complexes, thereby ensuring that cells remain in the G1 phase. G1 cyclins accumulate and phosphorylate Sic1, thereby making it a target for degradation (K�ivom�gi et al., 2011). The S cyclin-Cdk1 complex is therefore no longer inhibited and facilitates the initiation of S phase.
Cyclin-Dependent Kinase Regulation of Proteolysis
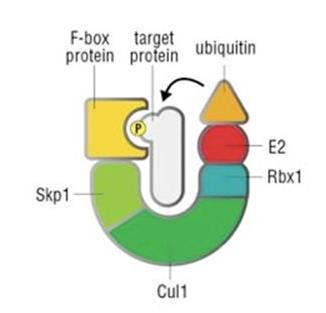
A major part of cell cycle control is the destruction of proteins used during previous phases. Cyclin-Cdks promote the ubiquitin-mediated proteolytic degradation of these proteins (Pagano, 2006; Benmaamar and Pagano, 2005). Proteins that are marked with ubiquitin are targeted for destruction by the proteasome. Attachment of ubiquitin to a protein requires the enzyme ubiquitin ligase. How is this process controlled? For many cell cycle regulators, recognition by ubiquitin ligase only occurs after they have been phosphorylated by cyclin-Cdk complexes. Cyclin-Cdks can also activate ubiquitin ligases by phosphorylating them.
One ubiquitin ligase is the Skp, Cullin, F-box containing, or SCF, complex (see simplified structure above) (Zheng et al., 2002). The SCF complex breaks down specific proteins that are phosphorylated by G1-S cyclin-Cdks, thereby controlling the G1-to-S phase transition (Ang and Harper, 2005). The F-box protein referred to in its name binds the protein that is to be targeted for ubiquitination. This is controlled by cyclin-Cdk phosphorylation. The budding yeast S phase cyclin-Cdk inhibitor Sic1 discussed previously is targeted by SCF-dependent destruction (Deshaies, 1999).
{kind=link}
Cdks can also directly activate a ubiquitin ligase called the anaphase-promoting complex (hereafter, APC) (Barford, 2011; Zachariae and Nasmyth, 1999). The APC is a complex machine, comprised of more than a dozen subunits (see diagram below). For an excellent review of the “unexpected multitude of mechanisms that control APC/C activity”, and the mechanism by which “this unusual ubiquitin ligase recognizes its substrates”, see Peters (2006). Inactivation of APC activity has been shown to prevent the cell from progressing through mitosis, causing lethality in all organisms so-far studied (Wirth et al., 2004). Jan-Michael Peters, in a 2006 paper in Nature (from which the above figure is excerpted), explains the enzyme’s mode of action:
Ubiquitin (Ub) is first activated and covalently bound through a thioester bond by the ubiquitin-activating (E1) enzyme and then transferred to the ubiquitin-conjugating (E2) enzyme with which the ubiquitin residue again forms a thioester bond. The ubiquitin-charged E2 enzyme interacts with anaphase promoting complex protein-11 (Apc11). This anaphase promoting complex/cyclosome (APC/C) subunit has ubiquitin ligase (E3) activity and promotes the transfer of the ubiquitin residue from the E2 enzyme to the substrate protein on which the C terminus of ubiquitin forms a covalent isopeptide bond with a lysine residue. In subsequent reactions, the attached ubiquitin can itself become ubiquitylated, resulting in the formation of a polyubiquitin chain…Substrates are recruited to the APC/C if they contain a D-box or a KEN-box. Both of these sequences are recognized by an APC/C co-activator, such as Cdh1 or Cdc20. Cdh1 binds to APC/C by interacting with two subunits, Cdc27 and Apc2. Cdc27 is one of several TPR proteins that are present in the APC/C…and Apc2 is a scaffold subunit that binds to Apc11 via a cullin domain. The small globular protein Doc1 is required for processive ubiquitylation of substrates and might also interact with the D-box of substrates, although direct evidence for such an interaction is lacking.
There are three major substrates that are a target of APC, two of which need to be targeted for destruction at the transition between metaphase and anaphase. Another target of the APC are the M cyclins, which need to be destroyed before the cell can exit mitosis. APC also has target substrates during the G1 phase. The APC differs from the SCF inasmuch as it lacks an F-box protein. Instead, its activator subunits determine substrate specificity. One activator subunit, Cdc20, targets proteins early in mitosis. Another, Cdh1, targets proteins late in mitosis and in G1. APC is activated early during mitosis by phosphorylation by M cyclin-Cdk, which promotes binding between the APC and Cdc20. This in turn promotes the destruction of a protein, called securin, that inhibits separation of the sister chromatids. The M cyclin-Cdk is in fact itself destroyed by APC-mediated ubiquitination that it was responsible for activating (Fung and Poon, 2005; Zachariae and Nasmyth, 1999). In yeast, M cyclin-Cdk also phosphorylates Cdh1. This ensures that Cdh1 becomes active only following anaphase, since the phosphate group is inhibitory, and is only removed when a phosphatase enzyme is activated after anaphase. Cdh1 then targets M cyclins for destruction. This allows the cell to exit mitosis and enter the G1 phase again. If M cyclins accumulated again, the cell would disastrously re-enter mitosis. Thus, Cdh1 remains active during the G1 phase. S cyclins will not accumulate, however, while Cdk1 is present. Cdk1 is thus inactivated by the G1 cyclins as they accumulate, thereby facilitating the transition from G1 to S phase.
The Role of Environmental Factors in Cell Cycle Control
As I mentioned in my previous article on the cell cycle, for many types of cells, commitment to another round of division (i.e. passing the restriction point) requires a critical cell size. In such cases, increase in cell mass can be promoted by growth factors and environmental cues, such as the presence of nutrients. For instance, in budding yeast, one of the environmental cues is an abundance of amino acids, which are transported into the cell. In animal cells, growth factors can bind growth factor receptors, which initiates a signaling cascade and culminates in the activation of a protein kinase called TOR (Kim and Guan, 2011). Upon activation of TOR, many processes are initiated, including an increased rate of protein synthesis, thereby driving an increase in cell mass.
How do cells of adult multicellular organisms know that a repair of tissue is required and thus they need to enter the cell cycle from the G0 state and begin dividing? Key to this process is another type of external signal called mitogens. These are small proteins that are secreted by adjacent cells and activate cyclin-Cdks, thereby signaling cell division. Some mitogens regulate only specific cell types, whereas others regulate many cell types. Epidermal growth factor (hereafter, EGF) is a mitogen that regulates division in many cell types — for instance, it has recently been shown to regulate hematopoietic stem cell regeneration following injury (Doan et al., 2013). Mitogens bind to a receptor that activates another signal transduction cascade involving a number of kinases, culminating in the activation of G1 and S genes. The G1 cyclin, cyclin D (discussed previously), associates with and activates Cdk4. Cdk4 is then able to phosphorylate Rb, freeing up E2F to activate the transcription of G1 genes.
The signaling cascade activated by mitogen receptor binding (called the MAP kinase, or MAPK, signaling pathway) is shown in the animation below:
Be sure to also check out this video for another worthwhile animation of the MAPK signaling pathway.
The Evolution of the Eukaryotic Cell Cycle
It is important to bear in mind that, while mammals possess multiple Cdks that are involved in cell cycle control, some other organisms (e.g. budding yeast) get by with only a single Cdk, Cdk1, which is activated by multiple phase-specific cyclins and phosphorylates many substrates. In fact, in budding yeast, more than 75 Cdk1 substrate-targets have been identified (Enserink and Kolodner, 2010). There is also only a single essential CAK (Cak1p) in yeast (Kaldis et al., 1998).
Moreover, since there is a significant degree of functional overlap between Cdks, Cdks can often substitute or compensate for one another when one has been deleted (Satyanarayana and Kaldis, 2009; Malumbres and Barbacid, 2005). The highly conserved Cdk1 (the only one present in yeast as mentioned above), which is needed in order to (among other things) prevent re-replication of DNA during S phase and is thus indispensable for cell division, is a notable exception (Diril et al., 2012; Blewett, 2007; Hochegger et al., 2007). It therefore seems to me that the diversity of Cdks that we see in mammals could well be the product of rounds of gene duplication and recruitment (Krylov et al., 2003). Critics would also be right to point out that organisms exhibit variations in cell cycle organization. For example, as mentioned previously, budding yeast lacks a G2 phase.
Drosophila embryos, for their first 13 cycles, lack both the G1 and G2 phases, and shuttle between replication and mitosis. There are thus simpler versions of the cell cycle that are possible. For a comparative review of cell cycle control across eukaryotes, see Harashima et al. (2013); Lichtenberg et al. (2007); and Dyczkowski and Vingron (2005).
Having thus taken care to fairly state the facts that bear most in the evolutionist’s favor, let us now turn to some problems. The cell cycle depends on many essential and interdependent parts, and it is clear that significant increases in cell cycle complexity require corresponding increases in regulatory control, in many cases involving many coordinated changes. The phase-specific cyclin molecules (that are necessary for activating the Cdks to phosphorylate specific substrates at different phases) seem to be far more difficult to explain by a step-wise mechanism, since their timely expression is required to drive the cell cycle forward. The various inhibitors involved in the process also seem to defy explanation by neo-Darwinian means. Indeed, mutations in these molecules can often result in cancer due to uncontrolled cell division.
The APC complex, which targets mitotic proteins for destruction, is also known to be essential, and its disruption leads to embryonic lethality, as well as very detrimental consequences in adults (e.g. acute liver failure) (Wirth et al., 2004). Yet, it is an extremely complex machine requiring very high levels of regulatory control to ensure the timeliness of its expression and activity (Peters, 2006).
Another issue is the myriad cases that exist, in cell cycle control systems, of proteins that regulate their own regulator. As previously discussed, the mitotic cyclin-Cdk in fact promotes its own demise by activating its own destroyer, the anaphase promoting complex (Zachariae and Nasmyth, 1999). But careful attention must be paid to the timing of APC expression, since untimely activity of the APC complex (too early or too late) could have devastating consequences.
As one ventures deeper into the eukaryotic cell cycle, the more complexity becomes apparent, and there is much that I have yet to touch on. For example, one of the most remarkable features of the cell cycle is the regulatory control and function of microtubule-associated motor proteins during mitosis, such as in the assembly, maintenance and positioning of the mitotic spindle; the promotion of poleward chromatid movement; chromosome separation; and much more. I will discuss this topic in further depth in a later article (for an appetizer, see Vanneste et al., 2011; and Moore and Wordeman, 2004). When all the complexity and functional inter-dependency is considered, it should give one cause to be, at the very least, intuitively suspicious. Of course, strikingly counterintuitive theories can often be correct, but in such cases a substantial burden of evidence lies with the defenders of such a counterintuitive theory. It seems that, in the case of neo-Darwinism, that burden of proof has yet to be met.
Conclusion
The regulatory control circuits of the cell cycle constitute one of the most beautiful and elegant processes in molecular biology. This short review has barely scratched the surface. At a later date I hope to continue to explore these remarkable mechanisms in detail. I plan to discuss the quality control checkpoint pathways that monitor the cell for damage as it progresses through the cell cycle phases. We shall see again, I think, that the more one understands about the cell cycle, the more one finds evidence of intelligent design.